���գ�CFDA����ˡ����WˎƷע�Է�ĸ﹤��������������Ҋ�壩�����c��ˎƷע�Թ����k������2007�棩��ȣ���������Щ��ͬ������Щ���c��
2015��11��6�գ���؞���䌍������Ժ�P�ڸĸ�ˎƷ�t�������u�����ƶȵ���Ҋ�������l��2015��44 ̖��������_չˎƷ�����S�ɳ������ƶ�ԇ�c�������ĸ�ˎƷע�Է���������PҪ��ʳƷˎƷ�O�ܿ�������ˡ�ˎƷ�����S�ɳ������ƶ�ԇ�c������������Ҋ�壩���͡����WˎƷע�Է�ĸ﹤��������������Ҋ�壩��������Ҋ��2015��11��20��ǰͨ�^����]��������ʳƷˎƷ�O�ܿ��֡�
һ����ˎ�Ķ��x��׃�_��
������Ҋ�壺����ˎƷ�İ�ȫ�L�U�̶ȣ���ˎƷ�֞���ˎ�ͷ���ˎ�ɴ����Σ�����ˎƷԭ���Ժ��·f�ԵIJ�ͬ������ˎ�Mһ���֞鄓��ˎ��������ˎ���������ڷ���ˎ�У�����������ˎ������r��ͬ���Mһ�����֞錦�������С�����δ����ˎƷ�ķ��ƣ�����������ˎƷ�ķ����Լ���������ˎƷ��Ո�������������ˎ��ָδ���Ї������������N�۵�ˎƷ�����������о���δ����ˎƷ�{�����ˎ��
��ˎƷע�Թ����k������2007�棩��ˎƷע����Ո������ˎ��Ո������ˎ��Ո���M��ˎƷ��Ո�����a����Ո����ע����Ո����ˎ��Ո����ָδ�����Ї����������N�۵�ˎƷ��ע����Ո����������ˎƷ��׃���͡���׃�oˎ;�����������m���Y��ˎƷע������ˎ��Ո�ij��������ˎ��Ո����ָ���a����ʳƷˎƷ�O�����������������е����Ї��Ҙ˜ʵ�ˎƷ��ע����Ո��
������ˎ����c��ˎ�O�y��׃��
��ˎ����棬�����ˡ�2.5�����֪���Գɷݵ����÷���������Ҏ����Ƅ�����
��ˎ�O�y�ڷ��棬2.4�������֪���Գɷݵ����m���Y���Ƅ��������]����ˎ�O�y�ڣ�������Ҋ�匦��ˎ�O�y�ڞ�3�꣬����2.5�������֪���Գɷݵ����÷���������Ҏ����Ƅ�����ˎ�O�y��Ҳ��3�ꡣ07��桶ˎƷע�Թ����k�����л��WˎƷ3.1�3.2�3.3�����������ˎ�O�y�ڣ����ǰ���������Ҋ��ϲ��M��3����ƾ������С�����δ���е�ˎƷ���o��ˎ�O�y�ڡ�
���η��������Ҋ��]���ἰ��ˎ��ռ�ڡ�
��1 ������Ҋ�廯�Wע���·��2007��桶ˎƷע�Թ����k�������� 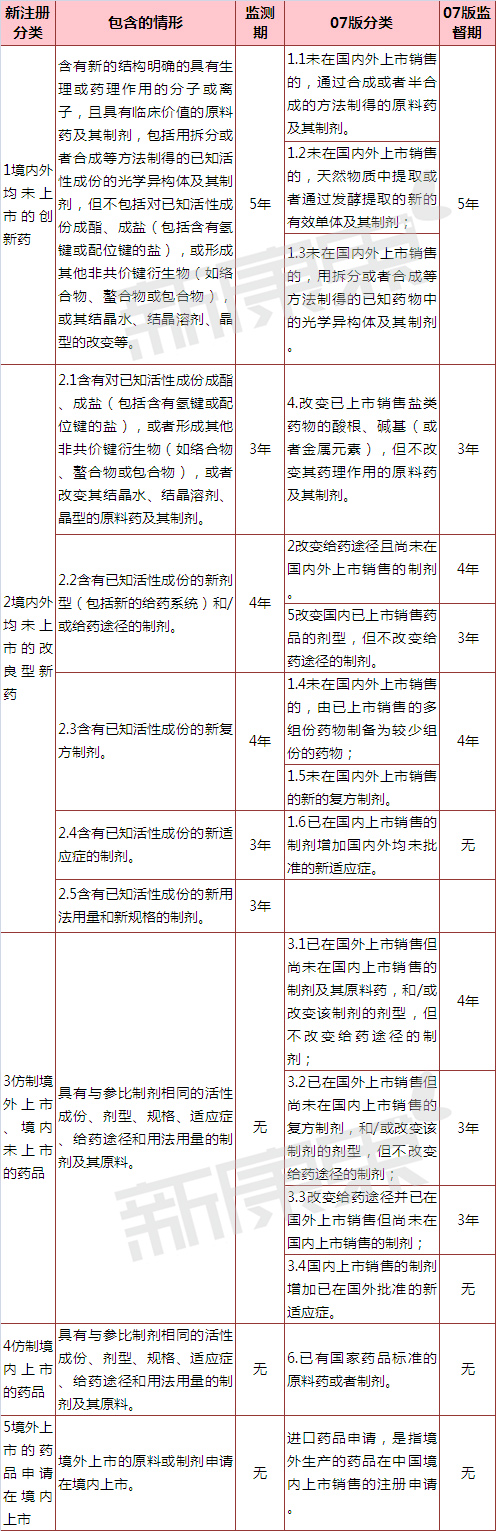 ע���˷����ֻ�ǂ��˳��У�һ����K����ԇ��Ҿ֞�ʡ�
�������WˎƷ��ע�Է������Y�ϸ�ע�ع�ˇ
�oՓ�DŽ���ˎ߀�Ƿ���ˎ�����WˎƷ��ע�Է������Y�ό����a��ˇ���^�̿��ơ����Ͽ��ơ��P�I���E�����g�w�Ŀ��ƶ������_��Ҫ������Y�������Ԕ�����о��Y�ϡ��������D�V���f�����_�l�A�Ό���Щ��ˇ���E�Ժ��|��ָ���M���˹�ˇ�l���ă��x�c�Ŵ��о����Գ���C��������ˇ���г���Č���֧�֣����C�F�е��о��c���a��Ҏģ�����Ͽ��ơ��������̼���Ҫ�O����ѻ����c�����aһ�£������a�������c�����aƷ���|�����ܷ�ӳ����ˇ�߂�һ�����جF�ԣ��M���C��ԓ���ˇ�ĺ������c�����a�Ŀ����ԡ����a��ˇ������Ԕ�Գ̶ȑ���ʹ�����I�ļ��g�ˆT�����������a��ˇ�����������؏����a�^�̣����Ƶ÷��Ϙ˜ʵĮaƷ�����⣬߀Ҫ�����r�ύ��ˇ�_�l�^�������a��ˇ����Ҫ׃���������������O�䡢��ˇ�����Լ���ˇ·���ȵ�׃������Ԕ���f�������P��֧������C�о��Y�ϡ��C������������Y�ϵ�Ҫ��Խ��Խ�ӵؚ⣬�����a�IJ���������
����Y��߀Ҫ��ˇ�u�rҪ�c������ԇ�������Ч��ԇ�Y���M�����P������Ҫ��R���аl�^���д��������Σ����������������R���о�������ԇ�Ŵ��������a�F���z��������ˇ��C���ȣ��Ę�Ʒ��r����������̖�����a�r�g�����c����Ҏģ����;�������ڷ�����ԇ���������Ч��ԇ�ȣ��������Y�����������P���|���ܳ����Լ�������Ҫ�|��ָ�ˣ����б����^�R��ԇ�/BEԇ��Ʒ��̎����ˇ�����a�O�䡢�������P�I��ˇ���E��ˇ�����Ŀ��ơ����g�w�Ŀ��Ƶ��c�M�������a���Į�ͬ�c���f���@Щ��Ƿ�Ӱ푮aƷ���|������Ҫ�f���о���r��
���Wˎˎ�W����Y��߀�����o������Y��Ҫ��Ͱ��b����/��������Y��Ҫ������������Ҋ�岢�oһ���������Դ�ע��˾�yһ�l���� |


